Introduction
This vignette walks through the DIAMOND RBH + Expansion clustering algorithm step by step, using real data from known fungal orthologs. Instead of describing algorithms conceptually, we show exactly what happens at each stage with actual sequence data.
The Dataset
We use 16 well-characterized proteins from three fungal species with known orthology relationships:
# Load the protein collection
clustering_dir <- system.file("testdata", "clustering", package = "paneffectR")
proteins <- readRDS(file.path(clustering_dir, "known_orthologs.rds"))
# Load expected relationships (ground truth)
expected <- read.csv(file.path(clustering_dir, "expected_orthogroups.csv"))
# Summary
proteins
#> -- protein_collection --
#> 3 assemblies, 16 total proteins
#>
#> # A tibble: 3 × 3
#> assembly_name n_proteins has_scores
#> <chr> <int> <lgl>
#> 1 ncra 5 FALSE
#> 2 scer 6 FALSE
#> 3 spom 5 FALSEThe dataset includes:
- 3 species: S. cerevisiae (budding yeast), S. pombe (fission yeast), N. crassa (bread mold)
- 5 conserved protein families: actin, GAPDH, histone H3, EF-1α, cytochrome c
- 1 species-specific singleton: mating pheromone MFA1 (only in S. cerevisiae)
expected |>
select(protein_id, assembly, expected_og, protein_name) |>
knitr::kable(caption = "Ground truth: expected orthogroup assignments")| protein_id | assembly | expected_og | protein_name |
|---|---|---|---|
| scer_ACT1 | scer | OG_actin | Actin |
| spom_act1 | spom | OG_actin | Actin |
| ncra_act | ncra | OG_actin | Actin |
| scer_TDH1 | scer | OG_gapdh | Glyceraldehyde-3-phosphate dehydrogenase 1 |
| spom_tdh1 | spom | OG_gapdh | Glyceraldehyde-3-phosphate dehydrogenase 1 |
| ncra_gpd1 | ncra | OG_gapdh | Glyceraldehyde-3-phosphate dehydrogenase |
| scer_HHT1 | scer | OG_histone_h3 | Histone H3 |
| spom_hht1 | spom | OG_histone_h3 | Histone H3.1/H3.2 |
| ncra_hh3 | ncra | OG_histone_h3 | Histone H3 |
| scer_TEF1 | scer | OG_ef1a | Elongation factor 1-alpha |
| spom_tef101 | spom | OG_ef1a | Elongation factor 1-alpha-A |
| ncra_tef1 | ncra | OG_ef1a | Elongation factor 1-alpha |
| scer_CYC1 | scer | OG_cyc | Cytochrome c isoform 1 |
| spom_cyc1 | spom | OG_cyc | Cytochrome c |
| ncra_cyc1 | ncra | OG_cyc | Cytochrome c |
| scer_MFA1 | scer | singleton | Mating factor alpha-1 |
Step 1: All-vs-All Alignment
The first step runs DIAMOND blastp to compare every protein against every other protein. This produces a table of sequence similarity hits.
paneffectR runs DIAMOND with a custom output format to get the columns needed for filtering and RBH detection:
diamond blastp -d db -q proteins.faa -o hits.tsv \
--outfmt 6 qseqid sseqid pident length qlen evalue bitscore
# Load pre-computed DIAMOND results
diamond_hits <- readRDS(file.path(clustering_dir, "diamond_hits.rds"))
# Show structure
cat("Total hits (excluding self-hits):", nrow(diamond_hits), "\n\n")
#> Total hits (excluding self-hits): 30
head(diamond_hits, 10) |>
knitr::kable(digits = 2, caption = "DIAMOND output with computed coverage")| qseqid | sseqid | pident | length | qlen | evalue | bitscore | qcov |
|---|---|---|---|---|---|---|---|
| ncra_act | spom_act1 | 92.0 | 375 | 375 | 0 | 716 | 100.00 |
| ncra_act | scer_ACT1 | 92.0 | 375 | 375 | 0 | 709 | 100.00 |
| ncra_gpd1 | spom_tdh1 | 69.1 | 333 | 338 | 0 | 463 | 98.52 |
| ncra_gpd1 | scer_TDH1 | 63.7 | 331 | 338 | 0 | 438 | 97.93 |
| ncra_hh3 | scer_HHT1 | 94.9 | 136 | 136 | 0 | 236 | 100.00 |
| ncra_hh3 | spom_hht1 | 92.6 | 136 | 136 | 0 | 235 | 100.00 |
| ncra_tef1 | spom_tef101 | 86.1 | 460 | 460 | 0 | 807 | 100.00 |
| ncra_tef1 | scer_TEF1 | 85.7 | 460 | 460 | 0 | 795 | 100.00 |
| ncra_cyc1 | spom_cyc1 | 70.4 | 108 | 108 | 0 | 175 | 100.00 |
| ncra_cyc1 | scer_CYC1 | 69.2 | 104 | 108 | 0 | 166 | 96.30 |
Understanding the columns
From DIAMOND directly (7 columns):
| Column | Description |
|---|---|
qseqid |
Query protein ID |
sseqid |
Subject (hit) protein ID |
pident |
Percent sequence identity |
length |
Alignment length (amino acids) |
qlen |
Query sequence length |
evalue |
E-value (statistical significance; lower = better) |
bitscore |
Bit score (higher = better match) |
Computed by paneffectR (1 column):
| Column | Formula | Purpose |
|---|---|---|
qcov |
(length / qlen) * 100 |
Query coverage percentage, used for filtering |
Note: This differs from the default BLAST tabular format, which
includes alignment coordinates (qstart, qend,
sstart, send) and gap information
(mismatch, gapopen). We request only the
columns needed for clustering.
Visualizing hit scores
# Get best hit between each pair
best_hits <- diamond_hits |>
group_by(qseqid, sseqid) |>
slice_max(bitscore, n = 1) |>
ungroup()
# Order proteins by protein family (expected_og) to reveal ortholog blocks
protein_order <- expected |>
arrange(expected_og, assembly) |>
pull(protein_id)
# Convert to long format for ggplot with family-based ordering
plot_data <- best_hits |>
mutate(
qseqid = factor(qseqid, levels = protein_order),
sseqid = factor(sseqid, levels = protein_order)
)
ggplot(plot_data, aes(x = sseqid, y = qseqid, fill = bitscore)) +
geom_tile() +
scale_fill_viridis_c(name = "Bitscore") +
theme_minimal() +
theme(axis.text.x = element_text(angle = 45, hjust = 1, size = 8),
axis.text.y = element_text(size = 8)) +
labs(x = "Subject", y = "Query",
title = "DIAMOND blastp hit scores (ordered by protein family)",
subtitle = "Diagonal blocks show orthologs with high similarity")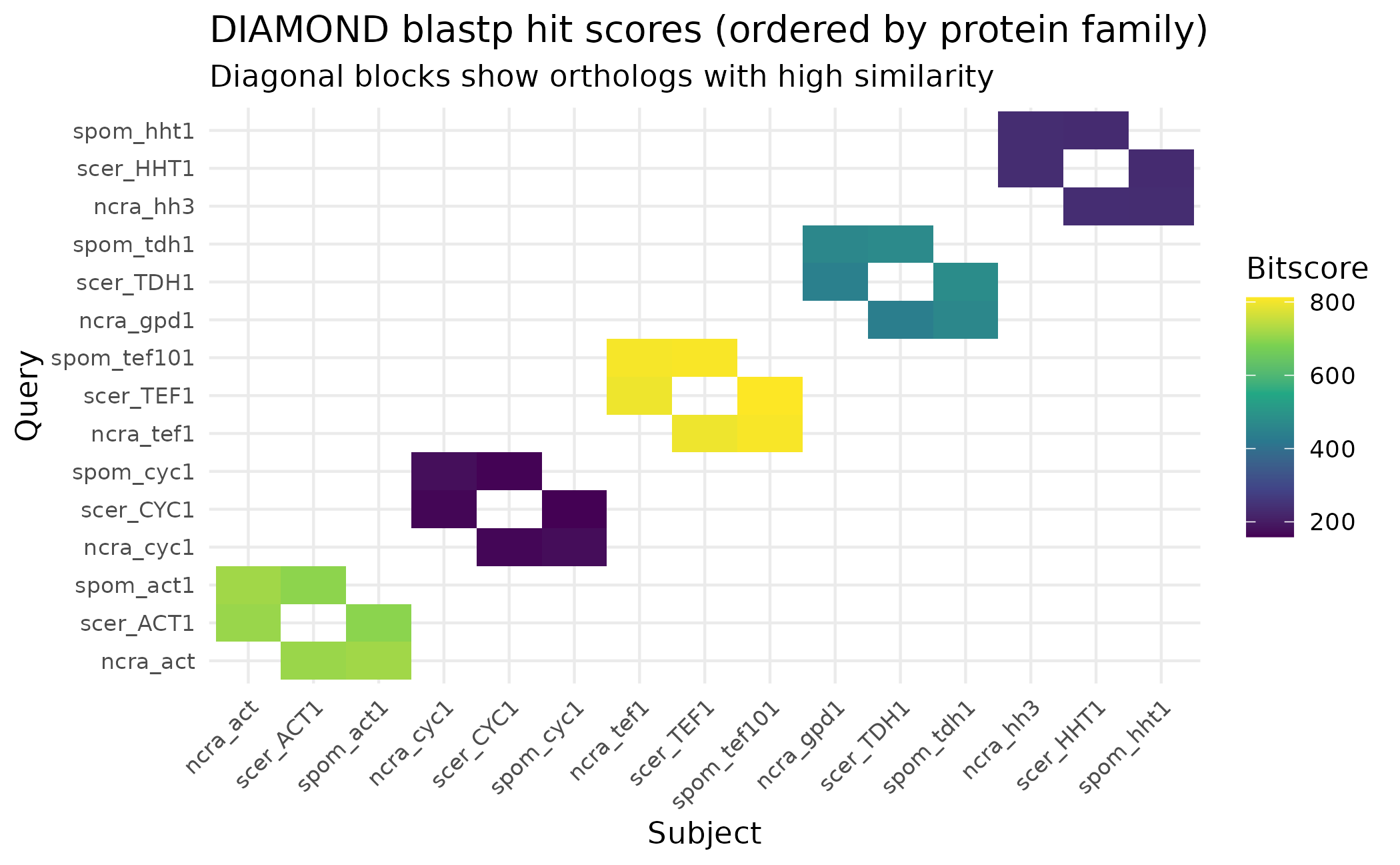
The heatmap reveals clear ortholog groups: proteins from the same family (e.g., all three actins) cluster together with high scores, while unrelated proteins show no hits.
Note: The singleton scer_MFA1 (mating
pheromone) is absent from this plot because it has no significant hits
to any other protein - exactly why it will end up as a singleton in the
final clustering.
Step 2: Filtering by Thresholds
Not all hits are meaningful. We apply thresholds to keep only significant matches:
| Parameter | Default | Purpose |
|---|---|---|
min_identity |
30% | Exclude very divergent matches |
min_coverage |
50% | Exclude partial matches |
evalue |
1e-5 | Exclude statistically insignificant matches |
# Load filtered hits
hits_filtered <- readRDS(file.path(clustering_dir, "diamond_hits_filtered.rds"))
cat("Before filtering:", nrow(diamond_hits), "hits\n")
#> Before filtering: 30 hits
cat("After filtering: ", nrow(hits_filtered), "hits\n")
#> After filtering: 30 hitsIn this case, all hits pass the filters because these are well-conserved proteins with high sequence similarity.
Step 3: Finding Reciprocal Best Hits
A reciprocal best hit (RBH) is when two proteins are each other’s best match. This is a stringent criterion for orthology:
- Find protein A’s best hit → B
- Find protein B’s best hit → back to A?
- If yes, A and B are reciprocal best hits
From our 30 filtered hits, paneffectR identified 5 RBH pairs:
rbh_pairs <- readRDS(file.path(clustering_dir, "rbh_pairs.rds"))
rbh_pairs |>
knitr::kable(caption = "Reciprocal best hit pairs identified")| protein_a | protein_b |
|---|---|
| ncra_act | spom_act1 |
| ncra_cyc1 | spom_cyc1 |
| ncra_hh3 | scer_HHT1 |
| scer_TDH1 | spom_tdh1 |
| scer_TEF1 | spom_tef101 |
Why not all orthologs form RBH pairs
Let’s trace through an example where RBH fails to connect obvious orthologs.
Question: Is scer_ACT1 (yeast actin) in
an RBH pair with ncra_act (N. crassa actin)?
Step 1: Find scer_ACT1’s best hit.
scer_act1_hits <- hits_filtered |>
filter(qseqid == "scer_ACT1") |>
arrange(desc(bitscore))
scer_act1_hits |>
select(qseqid, sseqid, pident, bitscore) |>
knitr::kable()| qseqid | sseqid | pident | bitscore |
|---|---|---|---|
| scer_ACT1 | ncra_act | 92.0 | 708 |
| scer_ACT1 | spom_act1 | 89.6 | 696 |
The best hit (highest bitscore) is ncra_act with score
708.
Step 2: Check the reverse - what is
ncra_act’s best hit?
ncra_act_hits <- hits_filtered |>
filter(qseqid == "ncra_act") |>
arrange(desc(bitscore))
ncra_act_hits |>
select(qseqid, sseqid, pident, bitscore) |>
knitr::kable()| qseqid | sseqid | pident | bitscore |
|---|---|---|---|
| ncra_act | spom_act1 | 92 | 716 |
| ncra_act | scer_ACT1 | 92 | 709 |
The best hit for ncra_act is spom_act1
(score 716), not scer_ACT1 (score
709).
Result: scer_ACT1 →
ncra_act, but ncra_act →
spom_act1. The relationship is not reciprocal, so they are
not an RBH pair.
This illustrates the limitation of pure RBH: scer_ACT1
is a clear actin ortholog, but because ncra_act is slightly
more similar to spom_act1, the yeast actin gets left out of
the RBH network. The expansion phase (Step 5) will rescue it.
Visualizing RBH pairs
With only 5 RBH pairs from 15 orthologous proteins, we’re clearly missing connections. To see the pattern, let’s visualize which species are linked by RBH for each protein family.
The figure below shows all 5 RBH pairs as lines connecting proteins across species. Each column is a protein family, each row is a species. Notice that RBH pairs only connect two species at a time - this is the fundamental limitation that the expansion phase addresses.
# Get protein positions for network layout
protein_positions <- expected |>
filter(expected_og != "singleton") |>
mutate(
x = as.numeric(factor(expected_og)),
y = as.numeric(factor(assembly, levels = c("ncra", "spom", "scer")))
)
# Plot
ggplot() +
# Add edges for RBH pairs
geom_segment(
data = rbh_pairs |>
left_join(protein_positions |> select(protein_id, x, y),
by = c("protein_a" = "protein_id")) |>
rename(x1 = x, y1 = y) |>
left_join(protein_positions |> select(protein_id, x, y),
by = c("protein_b" = "protein_id")) |>
rename(x2 = x, y2 = y),
aes(x = x1, y = y1, xend = x2, yend = y2),
color = "steelblue", linewidth = 1.5, alpha = 0.6
) +
# Add protein points
geom_point(data = protein_positions,
aes(x = x, y = y, color = assembly),
size = 4) +
geom_text(data = protein_positions,
aes(x = x, y = y, label = protein_id),
size = 2.5, vjust = -1) +
scale_y_continuous(
breaks = 1:3,
labels = c("N. crassa", "S. pombe", "S. cerevisiae")
) +
scale_x_continuous(
breaks = 1:5,
labels = c("actin", "cyc", "ef1a", "gapdh", "h3")
) +
scale_color_manual(values = c(ncra = "#E69F00", spom = "#56B4E9", scer = "#009E73")) +
theme_minimal() +
labs(title = "RBH pairs connect proteins across species",
subtitle = "Lines show reciprocal best hit relationships",
x = "Protein family", y = NULL) +
theme(legend.position = "none",
panel.grid.minor = element_blank())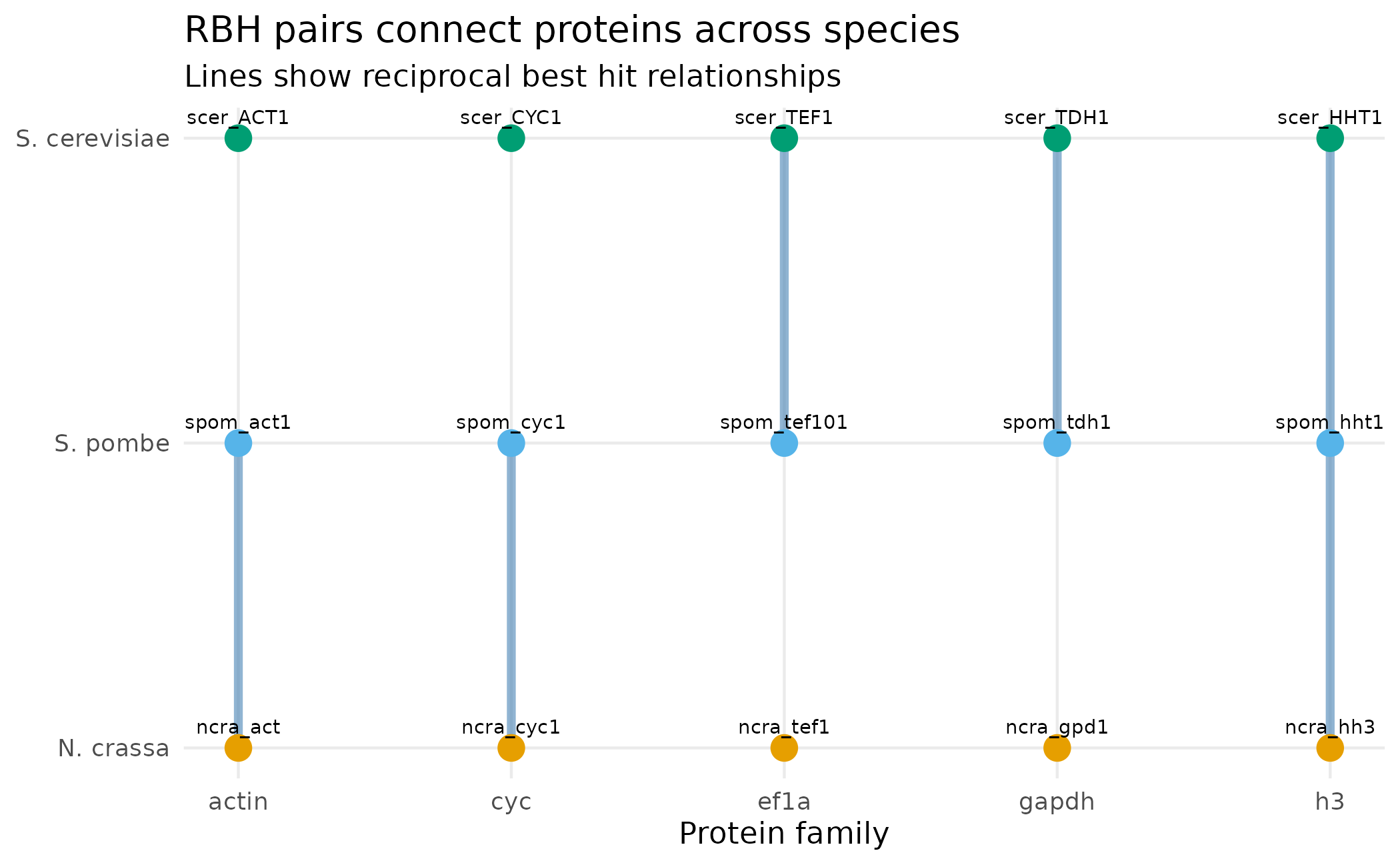
Notice that RBH pairs only connect two species at a time. This is the fundamental limitation of pure RBH: it can’t directly identify orthogroups spanning three or more species.
Step 4: Building Seed Clusters
The RBH pairs form the “seeds” for orthogroups. We find connected components in the RBH graph: proteins linked through any chain of RBH pairs form one group.
orthogroups_seed <- readRDS(file.path(clustering_dir, "orthogroups_seed.rds"))
orthogroups_seed |>
arrange(orthogroup_id, protein_id) |>
knitr::kable(caption = "Seed orthogroups from RBH connected components")| orthogroup_id | protein_id |
|---|---|
| OG0001 | ncra_act |
| OG0001 | spom_act1 |
| OG0002 | ncra_cyc1 |
| OG0002 | spom_cyc1 |
| OG0003 | ncra_hh3 |
| OG0003 | scer_HHT1 |
| OG0004 | scer_TDH1 |
| OG0004 | spom_tdh1 |
| OG0005 | scer_TEF1 |
| OG0005 | spom_tef101 |
After the seed phase:
- 5 orthogroups with 10 proteins (2 proteins each)
- 6 singletons: proteins not in any RBH pair
seed_summary <- orthogroups_seed |>
group_by(orthogroup_id) |>
summarize(n_proteins = n(), proteins = paste(protein_id, collapse = ", "))
seed_summary |>
knitr::kable(caption = "Seed orthogroup composition")| orthogroup_id | n_proteins | proteins |
|---|---|---|
| OG0001 | 2 | ncra_act, spom_act1 |
| OG0002 | 2 | ncra_cyc1, spom_cyc1 |
| OG0003 | 2 | ncra_hh3, scer_HHT1 |
| OG0004 | 2 | scer_TDH1, spom_tdh1 |
| OG0005 | 2 | scer_TEF1, spom_tef101 |
# List singletons
all_proteins_list <- unlist(lapply(proteins$assemblies, function(ps) ps$proteins$protein_id))
singletons <- setdiff(all_proteins_list, orthogroups_seed$protein_id)
cat("\nSingletons after seed phase:", paste(singletons, collapse = ", "), "\n")
#>
#> Singletons after seed phase: ncra_gpd1, ncra_tef1, scer_ACT1, scer_CYC1, scer_MFA1, spom_hht1The actins (scer_ACT1), GAPDH (ncra_gpd1),
cytochrome c (scer_CYC1), EF-1α (ncra_tef1),
and histone H3 (spom_hht1) are all singletons - they
weren’t part of any RBH pair even though they have clear orthologs!
Step 5: Expansion Phase
The expansion phase addresses the RBH limitation by adding singletons to existing orthogroups when their best hit is already a cluster member:
- For each singleton, find its best hit
- If that hit is in an orthogroup, add the singleton to that group
- Repeat until no more proteins can be added
The DIAMOND hits behind RBH pairs
First, let’s see the actual DIAMOND output rows that support the 5 RBH pairs. For each RBH pair, both directions must be best hits:
# Get the DIAMOND hits that form RBH pairs
# For each RBH pair, show both directions from the original DIAMOND output
rbh_evidence <- rbh_pairs |>
rowwise() |>
mutate(
# Get hit A -> B
hit_ab = list(hits_filtered |>
filter(qseqid == protein_a, sseqid == protein_b) |>
select(qseqid, sseqid, pident, bitscore)),
# Get hit B -> A
hit_ba = list(hits_filtered |>
filter(qseqid == protein_b, sseqid == protein_a) |>
select(qseqid, sseqid, pident, bitscore))
) |>
ungroup()
# Combine into one table
rbh_hits <- bind_rows(
bind_rows(rbh_evidence$hit_ab),
bind_rows(rbh_evidence$hit_ba)
) |>
arrange(qseqid, desc(bitscore))
rbh_hits |>
knitr::kable(
caption = "DIAMOND hits for RBH pairs (both directions)",
col.names = c("Query", "Subject", "% Identity", "Bitscore"),
digits = 1
)| Query | Subject | % Identity | Bitscore |
|---|---|---|---|
| ncra_act | spom_act1 | 92.0 | 716 |
| ncra_cyc1 | spom_cyc1 | 70.4 | 175 |
| ncra_hh3 | scer_HHT1 | 94.9 | 236 |
| scer_HHT1 | ncra_hh3 | 94.9 | 235 |
| scer_TDH1 | spom_tdh1 | 70.7 | 475 |
| scer_TEF1 | spom_tef101 | 87.1 | 812 |
| spom_act1 | ncra_act | 92.0 | 716 |
| spom_cyc1 | ncra_cyc1 | 70.4 | 177 |
| spom_tdh1 | scer_TDH1 | 70.7 | 470 |
| spom_tef101 | scer_TEF1 | 86.7 | 808 |
Each protein in an RBH pair has the other as its top hit (highest bitscore).
The DIAMOND hits behind expansion
Now let’s see the DIAMOND evidence for expansion. Each singleton’s best hit must be in an existing cluster:
expansion_trace <- readRDS(file.path(clustering_dir, "expansion_trace.rds"))
# For each singleton, show its DIAMOND hits (best hit first)
singletons_list <- expansion_trace$singleton
singleton_hits <- hits_filtered |>
filter(qseqid %in% singletons_list) |>
group_by(qseqid) |>
arrange(desc(bitscore)) |>
slice_head(n = 2) |> # Show top 2 hits for context
ungroup() |>
select(qseqid, sseqid, pident, bitscore)
# Add cluster membership info
clustered_proteins <- orthogroups_seed$protein_id
singleton_hits <- singleton_hits |>
mutate(
hit_in_cluster = ifelse(sseqid %in% clustered_proteins, "Yes", "No")
)
singleton_hits |>
knitr::kable(
caption = "DIAMOND hits from singletons (top 2 per singleton)",
col.names = c("Singleton", "Hit", "% Identity", "Bitscore", "Hit in cluster?"),
digits = 1
)| Singleton | Hit | % Identity | Bitscore | Hit in cluster? |
|---|---|---|---|---|
| ncra_gpd1 | spom_tdh1 | 69.1 | 463 | Yes |
| ncra_gpd1 | scer_TDH1 | 63.7 | 438 | Yes |
| ncra_tef1 | spom_tef101 | 86.1 | 807 | Yes |
| ncra_tef1 | scer_TEF1 | 85.7 | 795 | Yes |
| scer_ACT1 | ncra_act | 92.0 | 708 | Yes |
| scer_ACT1 | spom_act1 | 89.6 | 696 | Yes |
| scer_CYC1 | ncra_cyc1 | 69.2 | 165 | Yes |
| scer_CYC1 | spom_cyc1 | 70.2 | 159 | Yes |
| spom_hht1 | ncra_hh3 | 92.6 | 235 | Yes |
| spom_hht1 | scer_HHT1 | 92.6 | 232 | Yes |
For each singleton, the best hit (highest bitscore) is already in a seed cluster, so the singleton joins that cluster.
# Summarize the joins
expansion_trace |>
select(singleton, best_hit, best_hit_bitscore, target_cluster) |>
knitr::kable(
caption = "Expansion decisions: singleton joins cluster via best hit",
col.names = c("Singleton", "Best hit", "Bitscore", "Joins cluster"),
digits = 0
)| Singleton | Best hit | Bitscore | Joins cluster |
|---|---|---|---|
| ncra_gpd1 | spom_tdh1 | 463 | OG0004 |
| ncra_tef1 | spom_tef101 | 807 | OG0005 |
| scer_ACT1 | ncra_act | 708 | OG0001 |
| scer_CYC1 | ncra_cyc1 | 165 | OG0002 |
| spom_hht1 | ncra_hh3 | 235 | OG0003 |
Visualizing the clustering network
The network below shows how DIAMOND scores connect proteins. Edge thickness and labels show bitscores from the DIAMOND output. Blue edges are RBH pairs (seeds); orange edges show expansion connections.
library(igraph)
# Build edges from RBH pairs with their DIAMOND scores
rbh_with_scores <- rbh_pairs |>
left_join(
hits_filtered |> select(qseqid, sseqid, bitscore),
by = c("protein_a" = "qseqid", "protein_b" = "sseqid")
) |>
left_join(
hits_filtered |> select(qseqid, sseqid, bitscore),
by = c("protein_a" = "sseqid", "protein_b" = "qseqid"),
suffix = c("", "_rev")
) |>
mutate(bitscore = coalesce(bitscore, bitscore_rev)) |>
select(from = protein_a, to = protein_b, bitscore) |>
mutate(type = "RBH (seed)")
# Expansion edges already have scores
expansion_with_scores <- expansion_trace |>
select(from = singleton, to = best_hit, bitscore = best_hit_bitscore) |>
mutate(type = "Expansion")
# Combine all edges
all_edges <- bind_rows(rbh_with_scores, expansion_with_scores)
# Get node info (exclude singleton MFA1 which has no edges)
nodes <- expected |>
filter(expected_og != "singleton") |>
select(protein_id, assembly, expected_og)
# Create igraph object
g <- graph_from_data_frame(all_edges, directed = FALSE, vertices = nodes)
# Use Fruchterman-Reingold layout for natural clustering
set.seed(42)
layout <- layout_with_fr(g)
# Scale up to increase distance between nodes
layout <- layout * 2
layout_df <- as.data.frame(layout)
names(layout_df) <- c("x", "y")
layout_df$protein_id <- V(g)$name
# Add node attributes
layout_df <- layout_df |>
left_join(nodes, by = "protein_id")
# Prepare edge coordinates
edge_df <- all_edges |>
left_join(layout_df |> select(protein_id, x, y), by = c("from" = "protein_id")) |>
rename(x1 = x, y1 = y) |>
left_join(layout_df |> select(protein_id, x, y), by = c("to" = "protein_id")) |>
rename(x2 = x, y2 = y) |>
mutate(
xmid = (x1 + x2) / 2,
ymid = (y1 + y2) / 2,
score_label = round(bitscore)
)
# Plot
ggplot() +
# RBH edges (solid, fixed width)
geom_segment(
data = filter(edge_df, type == "RBH (seed)"),
aes(x = x1, y = y1, xend = x2, yend = y2),
color = "steelblue", linewidth = 1.5, alpha = 0.8
) +
# Expansion edges (dashed, fixed width)
geom_segment(
data = filter(edge_df, type == "Expansion"),
aes(x = x1, y = y1, xend = x2, yend = y2),
color = "darkorange", linewidth = 1.5, linetype = "dashed", alpha = 0.8
) +
# Protein nodes (large)
geom_point(
data = layout_df,
aes(x = x, y = y, color = assembly),
size = 14
) +
# Protein labels (inside nodes)
geom_text(
data = layout_df,
aes(x = x, y = y, label = protein_id),
size = 4.4, fontface = "bold", color = "black"
) +
# Edge score labels
geom_text(
data = edge_df,
aes(x = xmid, y = ymid, label = score_label),
size = 6, fontface = "bold"
) +
scale_color_manual(
values = c(ncra = "#E69F00", spom = "#56B4E9", scer = "#009E73"),
labels = c(ncra = "N. crassa", spom = "S. pombe", scer = "S. cerevisiae")
) +
coord_cartesian(clip = "off") +
expand_limits(x = c(min(layout_df$x) - 0.5, max(layout_df$x) + 0.5),
y = c(min(layout_df$y) - 0.5, max(layout_df$y) + 0.5)) +
theme_void() +
labs(
title = "Clustering network with DIAMOND bitscores",
subtitle = "Blue solid = RBH pairs (seed clusters), Orange dashed = expansion joins",
color = "Species"
) +
theme(
legend.position = "bottom",
plot.title = element_text(hjust = 0.5, face = "bold"),
plot.subtitle = element_text(hjust = 0.5)
)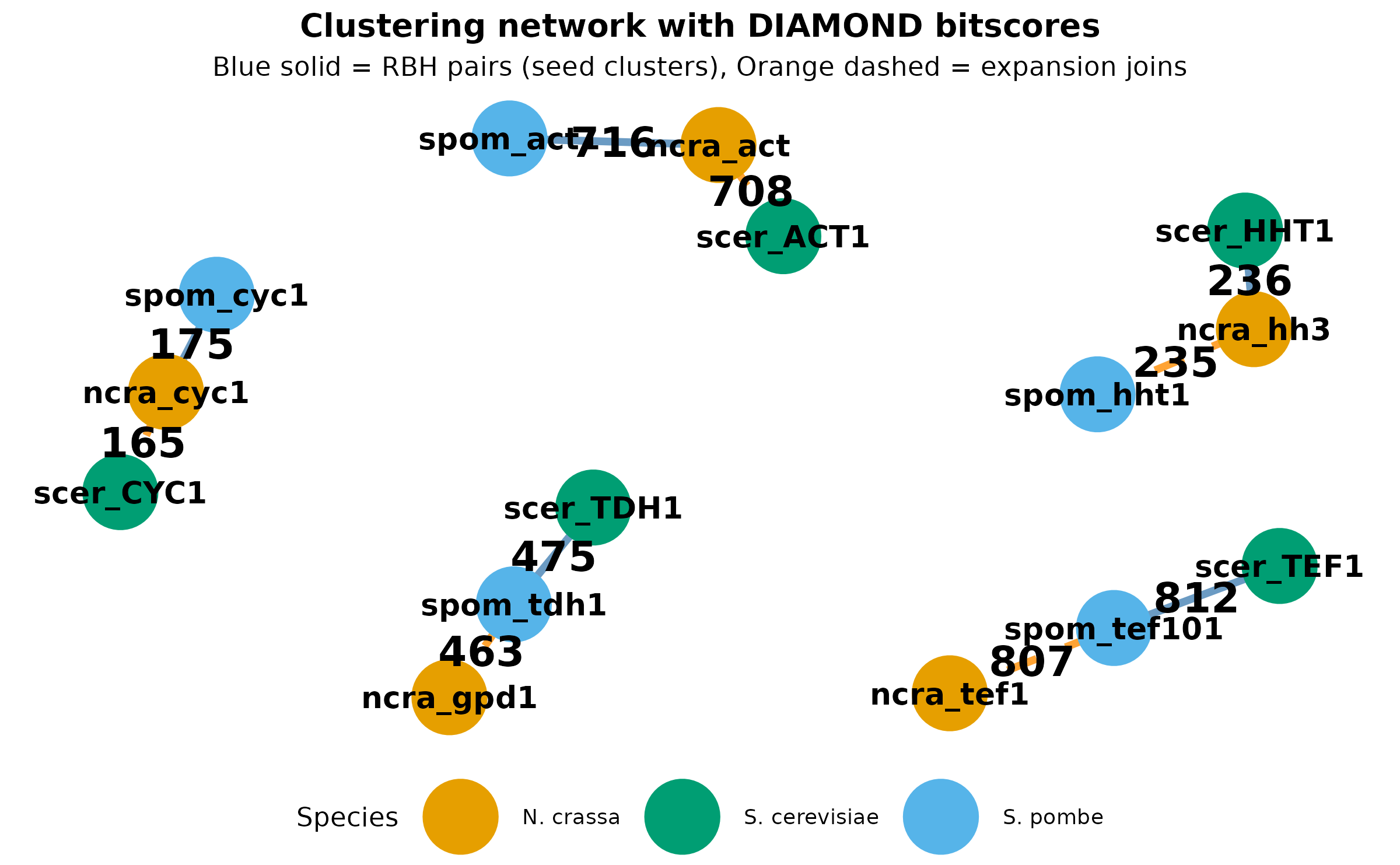
The network naturally clusters into 5 groups (the orthogroups). Proteins with high DIAMOND scores are pulled together. Notice how each orthogroup has 3 members connected by a mix of RBH (seed) and expansion edges.
What happened in expansion
Take scer_ACT1 as an example:
- It’s not in an RBH pair (its best hit
ncra_actprefersspom_act1) - But
ncra_actIS in OG0001 (via RBH withspom_act1) - So
scer_ACT1joins OG0001 through its best-hit connection
This “anchor and expand” strategy lets orthogroups grow beyond strict RBH pairs while maintaining the high-confidence RBH relationships as a foundation.
In iteration 1, all 5 “missing” orthologs found their way home:
# Show before and after with expected orthogroup names
orthogroups_expanded <- readRDS(file.path(clustering_dir, "orthogroups_expanded.rds"))
# Add expected orthogroup info
protein_info <- expected |>
select(protein_id, expected_og)
cat("=== Before Expansion (Seed Phase) ===\n")
#> === Before Expansion (Seed Phase) ===
orthogroups_seed |>
left_join(protein_info, by = "protein_id") |>
group_by(orthogroup_id) |>
summarize(
protein_family = first(expected_og),
members = paste(sort(protein_id), collapse = ", ")
) |>
knitr::kable()| orthogroup_id | protein_family | members |
|---|---|---|
| OG0001 | OG_actin | ncra_act, spom_act1 |
| OG0002 | OG_cyc | ncra_cyc1, spom_cyc1 |
| OG0003 | OG_histone_h3 | ncra_hh3, scer_HHT1 |
| OG0004 | OG_gapdh | scer_TDH1, spom_tdh1 |
| OG0005 | OG_ef1a | scer_TEF1, spom_tef101 |
cat("\n=== After Expansion ===\n")
#>
#> === After Expansion ===
orthogroups_expanded |>
left_join(protein_info, by = "protein_id") |>
group_by(orthogroup_id) |>
summarize(
protein_family = first(expected_og),
members = paste(sort(protein_id), collapse = ", "),
n = n()
) |>
knitr::kable()| orthogroup_id | protein_family | members | n |
|---|---|---|---|
| OG0001 | OG_actin | ncra_act, scer_ACT1, spom_act1 | 3 |
| OG0002 | OG_cyc | ncra_cyc1, scer_CYC1, spom_cyc1 | 3 |
| OG0003 | OG_histone_h3 | ncra_hh3, scer_HHT1, spom_hht1 | 3 |
| OG0004 | OG_gapdh | ncra_gpd1, scer_TDH1, spom_tdh1 | 3 |
| OG0005 | OG_ef1a | ncra_tef1, scer_TEF1, spom_tef101 | 3 |
# Check singleton
final_singletons <- setdiff(all_proteins_list, orthogroups_expanded$protein_id)
cat("\nFinal singleton:", final_singletons, "\n")
#>
#> Final singleton: scer_MFA1All 5 protein families now have their complete set of 3 orthologs,
and scer_MFA1 correctly remains a singleton.
Effect of Parameters
What happens with stricter thresholds? Let’s compare:
# Load strict parameter results
hits_strict <- readRDS(file.path(clustering_dir, "diamond_hits_strict.rds"))
orthogroups_strict <- readRDS(file.path(clustering_dir, "orthogroups_strict.rds"))
cat("Default parameters (min_identity = 30%):\n")
#> Default parameters (min_identity = 30%):
cat(" Hits passing filter:", nrow(hits_filtered), "\n")
#> Hits passing filter: 30
cat(" Orthogroups:", length(unique(orthogroups_expanded$orthogroup_id)), "\n")
#> Orthogroups: 5
cat(" Proteins in orthogroups:", nrow(orthogroups_expanded), "\n")
#> Proteins in orthogroups: 15
cat("\nStrict parameters (min_identity = 70%):\n")
#>
#> Strict parameters (min_identity = 70%):
cat(" Hits passing filter:", nrow(hits_strict), "\n")
#> Hits passing filter: 24
cat(" Orthogroups:", length(unique(orthogroups_strict$orthogroup_id)), "\n")
#> Orthogroups: 5
cat(" Proteins in orthogroups:", nrow(orthogroups_strict), "\n")
#> Proteins in orthogroups: 14Which proteins are affected?
# Find proteins that got excluded
proteins_strict <- orthogroups_strict$protein_id
proteins_default <- orthogroups_expanded$protein_id
newly_singletons <- setdiff(proteins_default, proteins_strict)
cat("Proteins excluded by strict parameters:\n")
#> Proteins excluded by strict parameters:
expected |>
filter(protein_id %in% newly_singletons) |>
select(protein_id, protein_name, expected_og) |>
knitr::kable()| protein_id | protein_name | expected_og |
|---|---|---|
| ncra_gpd1 | Glyceraldehyde-3-phosphate dehydrogenase | OG_gapdh |
# Show the excluded hits
hits_filtered |>
filter(!qseqid %in% proteins_strict | !sseqid %in% proteins_strict) |>
filter(qseqid %in% newly_singletons | sseqid %in% newly_singletons) |>
select(qseqid, sseqid, pident, bitscore) |>
distinct() |>
knitr::kable(caption = "Hits excluded by 70% identity threshold")| qseqid | sseqid | pident | bitscore |
|---|---|---|---|
| ncra_gpd1 | spom_tdh1 | 69.1 | 463 |
| ncra_gpd1 | scer_TDH1 | 63.7 | 438 |
| scer_TDH1 | ncra_gpd1 | 63.7 | 441 |
| spom_tdh1 | ncra_gpd1 | 69.1 | 461 |
The GAPDH and cytochrome c families have cross-species identities around 63-69%, below the strict 70% threshold. These proteins become incorrectly isolated as singletons with overly strict parameters.
Distance Metrics
Once orthogroups are assigned, we can build a presence/absence matrix (rows = orthogroups, columns = assemblies). To compare how similar two assemblies are based on their shared orthogroup content, we need a distance metric. This is used for hierarchical clustering and visualizations like dendrograms.
Jaccard Distance (Recommended)
The Jaccard index measures similarity as the size of the intersection divided by the size of the union. The distance is 1 minus this similarity, ignoring shared absences:
# Load example data from visual testdata
visual_dir <- system.file("testdata", "visual", package = "paneffectR")
pa <- readRDS(file.path(visual_dir, "pa_binary.rds"))
plot_dendro(pa, distance_method = "jaccard") +
ggtitle("Assembly clustering with Jaccard distance")
Other distance options
Binary (Euclidean): Square root of the number of orthogroups where two assemblies differ.
Bray-Curtis: Originally designed for abundance data, but works with binary. See Wikipedia.
| Method | Best for |
|---|---|
jaccard |
Presence/absence data (default) |
binary |
When total differences matter more than proportions |
bray |
Abundance or score data |
Hierarchical Clustering Methods
After computing distances, assemblies are clustered hierarchically:
| Method | Behavior |
|---|---|
complete |
Conservative - maximizes distances between clusters |
average |
Balanced - common in phylogenetics |
ward.D2 |
Creates distinct, equal-sized clusters |
# Compare methods
plot_dendro(pa, cluster_method = "complete")
plot_dendro(pa, cluster_method = "average")
plot_dendro(pa, cluster_method = "ward.D2")Handling Singletons
Singletons are proteins not assigned to any orthogroup. They may represent:
- True unique genes: Horizontally transferred, species-specific
- Divergent homologs: Too different to pass similarity thresholds
- Artifacts: Mis-predicted ORFs, fragmented assemblies
# Using the visual test data
visual_dir <- system.file("testdata", "visual", package = "paneffectR")
clusters <- readRDS(file.path(visual_dir, "clusters_visual.rds"))
# Compare with and without singletons
pa_with <- build_pa_matrix(clusters, exclude_singletons = FALSE)
pa_without <- build_pa_matrix(clusters, exclude_singletons = TRUE)
cat("With singletons:", nrow(pa_with$matrix), "orthogroups\n")
#> With singletons: 62 orthogroups
cat("Without singletons:", nrow(pa_without$matrix), "orthogroups\n")
#> Without singletons: 50 orthogroupsWhen to include singletons: - Analyzing unique gene content - Calculating pan-genome size - Studying accessory genome
When to exclude singletons: - Focusing on shared variation - Phylogenetic analysis - Reducing noise
Future Additions
The cluster_proteins() function is designed to support
multiple clustering backends. Currently only DIAMOND RBH is implemented,
but the interface accepts a method parameter for future
expansion:
cluster_proteins(proteins, method = "diamond_rbh") # Current default
cluster_proteins(proteins, method = "orthofinder") # Planned
cluster_proteins(proteins, method = "mmseqs2") # PlannedOrthoFinder
OrthoFinder infers orthogroups using gene trees and is considered a gold standard for orthology inference. It handles paralogs more rigorously than RBH-based methods but requires more computation time.
MMseqs2
MMseqs2 provides ultra-fast protein clustering suitable for very large datasets. Its greedy clustering approach trades some sensitivity for speed, making it useful when analysing hundreds of assemblies.